预测地理人口使用基因组变异和k - means


火花峰会将于2016年6月6 - 8在旧金山。检查排满日程得到你的票
这篇文章从黛博拉·西格尔从西北与丹尼基因组中心和华盛顿大学的李从砖在他们合作的基因组变异分析亚当和火花。
1。基因组测序的概括
2。并行基因组变异分析
3所示。预测地理人口使用基因组变异和k - means
介绍
在过去的几年里,我们看到了一个基因组测序的快速减少成本和时间。潜在的理解基因组序列的变化范围从帮助我们识别人倾向于常见疾病,解决罕见疾病,使临床医生能够个性化个人处方和剂量。
在这个分三部分的博客,我们将提供一个基因组测序引物及其潜力。我们将关注基因组变异分析,基因组序列之间的差异,以及如何加速利用Apache火花和亚当(基因组处理一个可扩展的API和CLI)使用砖Community Edition。最后,我们将执行一个k - means聚类算法对基因组变异数据,建立一个模型,将预测个人的地理原产地种群基于这些变体。
这篇文章将集中在预测地理人口使用基因组变异和k - means。你也可以检查复习基因组测序的概括或更多的细节并行基因组变异分析。
预测地理人口使用基因组变异和k - means
我们将展示基因组变异分析进行k - means亚当数据使用Apache火花砖社区版。笔记本显示如何执行一个公共的数据分析1000人基因组计划使用大数据基因组学亚当项目(0.19.0版本)。我们尝试k - means聚类预测地理人口每人从和可视化结果。
准备
如同大多数数据科学项目,有许多准备的任务必须完成。在本例中,我们将从我们的示例展示笔记本:
- 将一个示例VCF文件转换为亚当镶花的格式
- 加载一组文件,描述了数据样本内的VCF /亚当拼花
- 亚当的数据读入抽样并开始并行处理的基因型
创建亚当铺文件
创建一个从VCF亚当铺文件,我们将首先加载使用亚当的VCF文件SparkContext loadGenotypes方法。通过使用adamParquetSave方法,我们保存VCF亚当拼花格式。
val gts:抽样(基因型)= sc.loadGenotypes (vcf_path)
gts.adamParquetSave (tmp_path)
加载板文件
虽然VCF样本数据包含id,它们不包含我们想要预测人口代码。虽然我们正在做一个无监督算法分析,我们仍然需要响应变量为了过滤我们的样品,估计我们的预测误差。我们可以得到人口为每个样本代码integrated_call_samples_v3.20130502.ALL.panel板文件从1000人基因组计划。
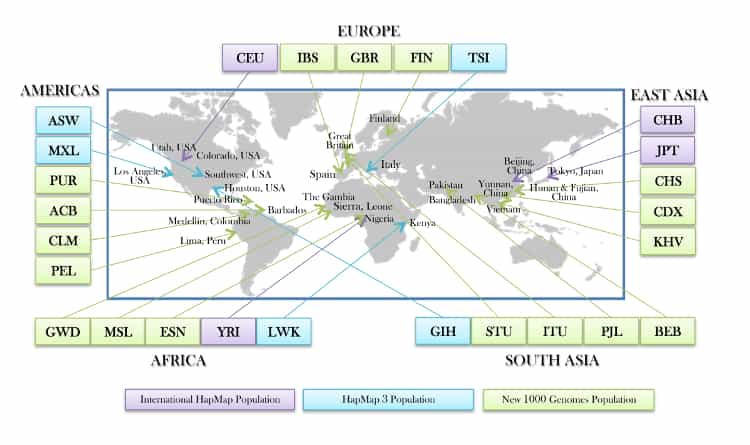
来源:1000 -基因组map_11 _750.jpg——6 - 12 - 2
下面的代码片段使用CSV加载板文件读者创造火花面板引发DataFrame。
val面板= sqlContext.read
.format (“com.databricks.spark.csv”)
.option(“头”,“真正的”)
.option (“inferSchema”,“真正的”)
.option(“分隔符”、“\ \ t”)
.load (panel_path)
对于我们的k - means聚类算法,我们将模型3集群,所以我们将创建一个过滤器3人口:英国从英格兰和苏格兰(GBR),非洲裔美国西南部(ASW),和汉族在北京,中国(慢性乙肝)。我们将通过创建一个filterPanelDataFrame只有这三个群体。这是一个小板,我们也广播给所有的执行人将导致更少的数据移动,当我们做进一步的操作,因此它将更有效率。
/ /创建过滤面板的三个人口
val filterPanel =面板。选择(“样例”、“流行”)。的地方(“流行(GBR,反潜战,慢性乙肝)”)
/ /取过滤板和广播
val fpanel = filterPanel
.rdd
. map {x = > x (0)。toString - > x (1) .toString}
.collectAsMap ()
val bPanel = sc.broadcast (fpanel)
并行处理的基因型
使用下面的命令,我们将加载我们的三个群体的基因型。可以更有效地并行,因为过滤板加载到内存中,播放所有节点(即bPanel)而拼花文件包含基因型数据使谓词下推到文件的水平。因此,只有我们感兴趣的记录从文件加载。
/ /创建过滤面板的三个人口
val popFilteredGts:抽样(基因型)= sc.loadGenotypes (tmp_path)。过滤器(基因型= > {bPanel.value.contains (genotype.getSampleId)})
笔记本包含一些额外的步骤包括:
- 勘探的数据——我们的数据已经6号染色体变异的一个小子集覆盖约一百万个碱基对。
- 清洗和过滤的数据缺失的数据或者变体triallelic。
- k - means聚类数据的准备——创建一个毫升向量为每个样本的变异(包含完全相同的顺序),然后退出运行模型对特征向量。
最终,805个变异的基因型中我们已经离开数据将被我们用来预测地理人口的特性。我们的下一个步骤是创建一个特征向量和DataFrame k - means聚类。
运行KMeans集群
使用上面的准备步骤,对基因组序列数据运行k - means聚类相似k - means例子在火花编程指南。
进口org.apache.spark.mllib.clustering。{KMeans, KMeansModel}
/ /使用KMeans集群数据分为三个类
val numClusters = 3
val numIterations = 20
val集群:KMeansModel = KMeans。火车(特性、numClusters numIterations)
现在,我们有了自己的模型——集群——让我们预测人口和计算混淆矩阵。首先,我们执行的任务创建predictionRDD其中包含原始值(即指原始的人口地理位置,反潜战,和GBR)和利用clusters.predictgeo的输出模型的预测基于特征(即基因组变异)。接下来,我们将进入predictDFDataFrame使其更容易查询(例如使用显示()命令,运行R命令在随后的细胞,等等)。最后,我们一起回filterPanel获取原始的标签(实际人口地理位置)。
/ /创建predictionRDD,利用集群。输出模型的预测方法预测地理位置
val predictionRDD:抽样(String、Int)] = dataPerSampleId。地图(sd = > {
(sd。_1,clusters.predict (sd._2))
})
/ /转换为DataFrame更容易查询数据
val predictDF = predictionRDD.toDF(“样本”,“预测”)
/ /加入回filterPanel得到原始的标签
val resultsDF = filterPanel。加入(predictDF“样本”)
/ /注册为临时表
resultsDF.registerTempTable (“results_table”)
/ /显示结果
显示器(resultsDF)
下面是输出的图形表示形式之间的预测价值和实际价值。
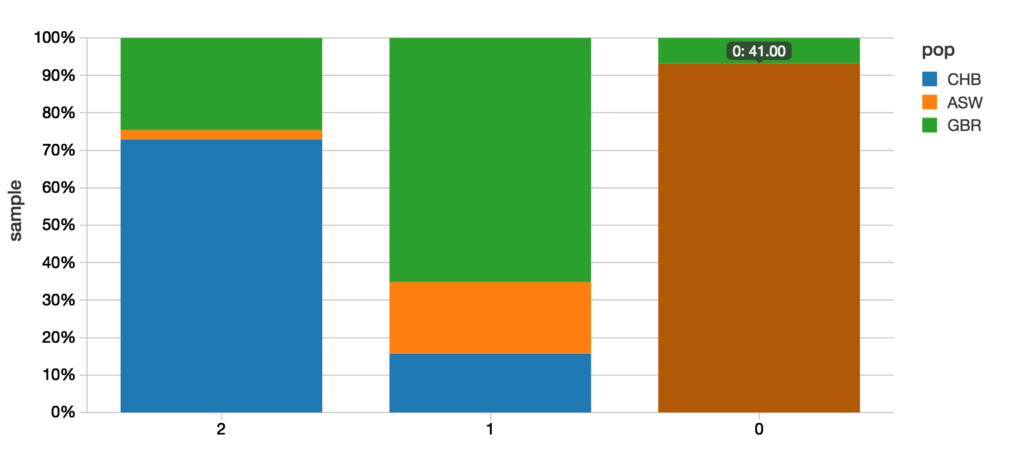
如何计算一个简单的例子混淆矩阵是使用r .虽然这笔记本主要是写在Scala中,我们可以添加一个新的细胞使用%表明我们正在使用r为我们的查询语言。
r %
resultsRDF prediction_pop慢性乙肝GBR反潜战
1 2 89年30 3
2 1 14 58 17
3 0 0 3 41
在笔记本,还有额外的SQL代码加入原样品,地理人口,人口代码,代码,这样你就可以预测地图预测个人样本。
集群可视化Lightning-Viz力量图
一个有趣的方法来可视化这些图通过k - means聚类是使用力量Lightning-Viz。笔记本包含Python代码用于创建闪电可视化。在下面的gif动画中,你可以看到三个集群代表三个人口(左上:2,右上:1,底部:0),预测的集群成员集群的顶点,而不同的颜色代表的人口。点击显示了人口sampleID、颜色(实际人口)和预测人口(顶点)行。

讨论
在这篇文章中,我们提供了一个基因组测序引物(基因组测序的概括)和周围的复杂性变异分析(并行基因组变异分析)。通过引入亚当,我们可以在一个分布式并行的过程变量的方式显著提高性能和精度分析。这已经证明在基因组变异分析进行k - means亚当数据使用Apache火花的笔记本,你可以为自己在运行砖社区版。基因组变异分析的承诺是,我们可以确定个体倾向于常见疾病,解决罕见疾病,提供个性化的治疗。正如我们已经看到了巨大的下降和大规模并行测序成本和时间,大规模并行处理可再生的生物信息学分析将帮助我们不断上涨的洪水序列的分析数据,甚至可能导致的分析方法,目前无货。最终,它将有助于医学的进展。
归因
我们想要给一个特定的电话下面的资源帮助我们创建了笔记本
- 大数据基因组学亚当项目
- 亚当:基因组学云规模计算格式和处理模式(伯克利AMPLab)
- 安迪•彼得雷拉闪电快基因组学与火花和亚当和相关的GitHub回购。
- 尼尔•弗格森用深度学习人口分层分析基因组数据。
- 马修ConlenLightning-Viz项目。
- 盖Danford SlideShare演示(基因组学与火花)
- 孟德尔基因组学中心发现数以百计的罕见的基因基础条件
- 国家卫生研究院基因组测序计划目标共同的基因基础,罕见的疾病
- 1000人基因组计划
,我们想感谢额外贡献和评论由安东尼·约瑟夫Xiangrui孟,Hossein Falaki,蒂姆·亨特。