简化基因组学管道与砖三角洲的规模


2019年3月7日, 在工程的博客
在砖试试这个笔记本
这个博客是第一个博客在我们的“大规模基因组学分析”系列。在本系列中,我们将演示如何砖统一为基因组学分析平bob体育亚洲版台bob体育客户端下载使客户能够分析人口规模的基因组数据。从我们的输出基因组学管道,本系列将提供一个教程使用砖质量控制运行样例,关节型,群体质量控制和高级统计遗传学分析。
因为完成人类基因组计划2003年,爆炸在数据受DNA测序的成本大幅下降,从3美元1今天第一个基因组在1000美元以下。
[1]人类基因组计划是一个美元3 b项目由美国能源部和美国国立卫生研究院始于1990年,完成于2003年。
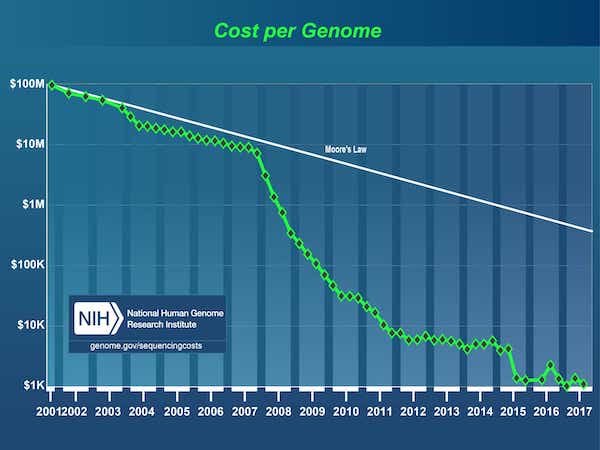
来源:DNA测序成本:数据
因此,基因组学领域已经成熟阶段,企业已开始在人口规模的DNA测序。然而,测序DNA代码仅仅是第一步,然后需要的原始数据转换为格式适用于分析。这通常是通过一系列的粘合在一起生物信息学工具使用自定义脚本和处理在单个节点上的数据,一次一个样本,直到我们最终与基因组变异的集合。生物信息学今天科学家们把大多数时间花在构建和维护这些管道。基因组数据集已经扩展到pb级别的时候,甚至已经成为具有挑战性的回答以下简单的问题及时:
- 这个月我们有多少样本测序吗?
- 独特的变异检测的总数是什么?
- 我们看到了多少变异在不同类别的变化?
进一步加剧这一问题,从成千上万的个人不能存储数据,跟踪和版本,同时保持可访问和可查询。因此,研究人员经常重复的基因组数据的子集在执行他们的分析,导致整体存储足迹和成本升级。为了缓解这个问题,今天的研究人员使用“数据冻结”的策略,通常六个月到两年之间,他们暂停了新数据,而是专注于冷冻现有数据的副本。没有解决方案,逐步建立在较短的时间框架分析,导致研究进展慢下来。
有一个引人注目的需要健壮的软件,可以使用基因组数据在工业规模,同时保持灵活性,科学家研究数据,进行迭代分析管道,并获得新的见解。
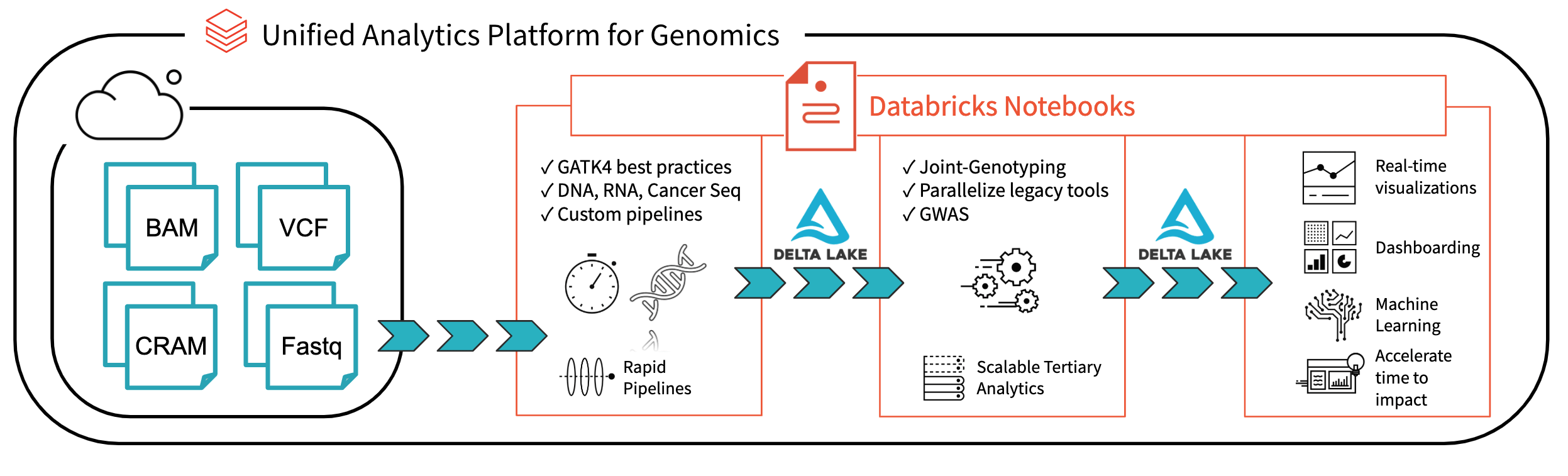
图1所示。端到端架构与砖基因组学分析
与砖三角洲:一个统一的实时大数据分析管理系统,砖平台已经迈出重要的一步解决数据bob体育客户端下载治理、数据访问和数据分析研究人员今天所面临的问题。与砖三角洲湖,你可以将你所有的基因组数据存储在一个地方,并创建分析,实时更新新的数据摄取。在我们结合优化bob体育亚洲版统一为基因组学分析平台bob体育客户端下载(UAP4G)阅读,写作,和处理基因组学文件格式,我们提供一个端到端的解决方案基因组学管道工作流。UAP4G架构提供了灵活性,允许顾客插入自己的管道和开发自己的高等分析。作为一个例子,我们强调以下指示板显示质量控制指标和可视化,可以计算并提出了一个自动时尚和定制以满足您的特定需求。
https://www.youtube.com/watch?v=73fMhDKXykU
在这个博客的其余部分,我们将介绍我们采取的措施是建立上述质量控制仪表板,实时更新为样本完成处理。通过使用一个Delta-based管道处理基因数据,我们的客户现在可以操作自己的管道,提供实时、连续取样可见性。与砖笔记本(和GitHub等集成和MLflow)他们可以跟踪和版本分析的方式将确保他们的结果是可再生的。他们bioinformaticians可以花更少的时间来维护管道和花更多的时间发现。我们看到UAP4G为引擎,将推动转变特别分析基因组学工业规模生产,使更好的洞察基因和疾病之间的联系。
读取样本数据
让我们开始通过阅读变异数据从一个小的样本;以下语句读取的数据为一个特定的sampleId使用砖三角洲格式并保存它(在delta_stream_output文件夹中)。
spark.read。\格式(“铺”)。\负载(“dbfs: / annotations_etl_parquet sampleId = "+“SRS000030_SRR709972”)。\写。\格式(“δ”)。\保存(delta_stream_outpath)注意,annotations_etl_parquet文件夹包含注释产生的1000基因组数据集存储在拼花格式。这些注释的ETL和处理进行使用砖的统一为基因组学分析平bob体育亚洲版台bob体育客户端下载。
砖三角洲开始流表
下面的声明中,我们创建的外显Apache火花DataFrame读取流(通过readStream)使用砖三角洲格式的数据。这是一个连续运行或动态DataFrame,即外显DataFrame加载新数据作为数据写入delta_stream_output文件夹。查看DataFrame外,我们可以运行DataFrame查询找到sampleId变异数的分组。
#读取的数据= spark.readStream外。格式(“δ”).load (delta_stream_outpath)#通过DataFrame查询显示数据显示器(exomes.groupBy (“sampleId”).count () .withColumnRenamed (“数”,“变异”))当执行显示声明中,砖的笔记本提供流媒体仪表板监控流工作。立即流工作的结果显示以下声明(即由sample_id变种)的计数。
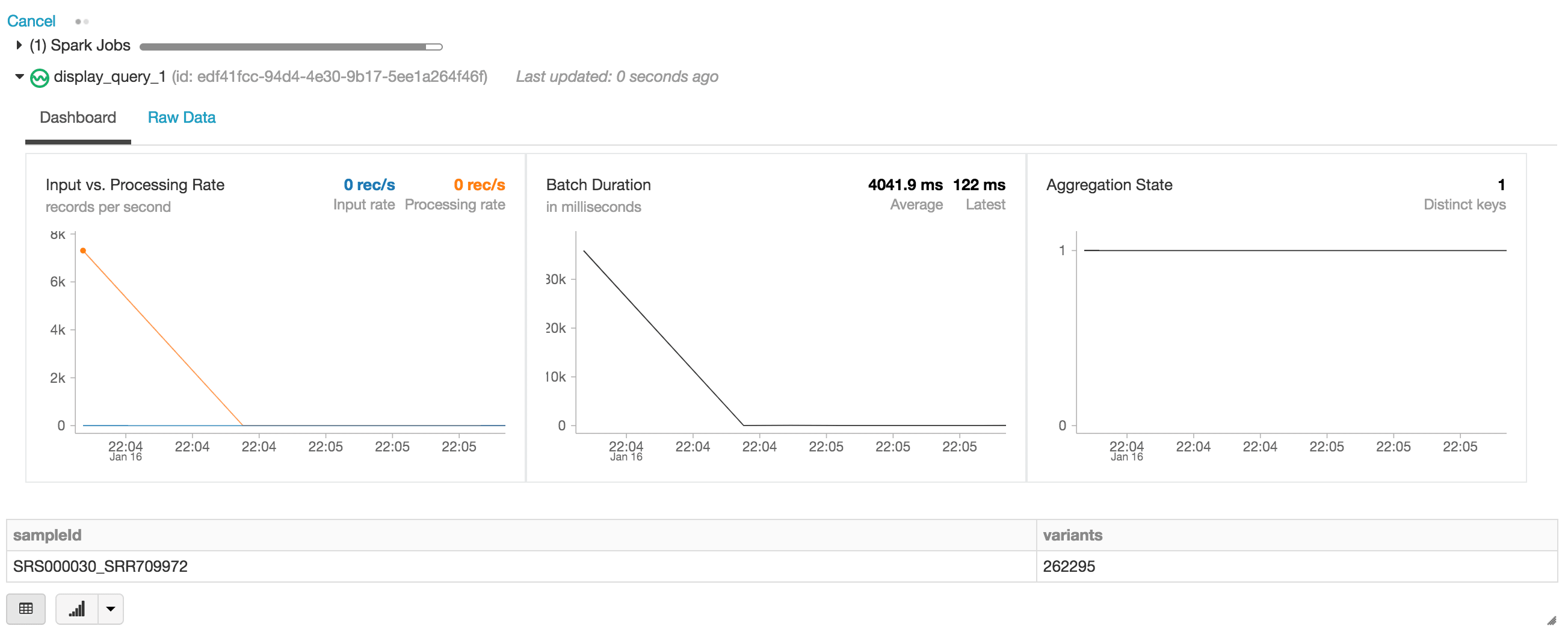
让我们继续回答最初的问题通过运行其他DataFrame根据我们查询外显DataFrame。
单核苷酸变异数
继续这个示例中,我们可以快速计算的数量单核苷酸变异(SNVs),如下图中显示。
%sql选择referenceAllele alternateAllele,数(1)作为GroupCount从snvs集团通过referenceAllele, alternateAllele订单通过GroupCountdesc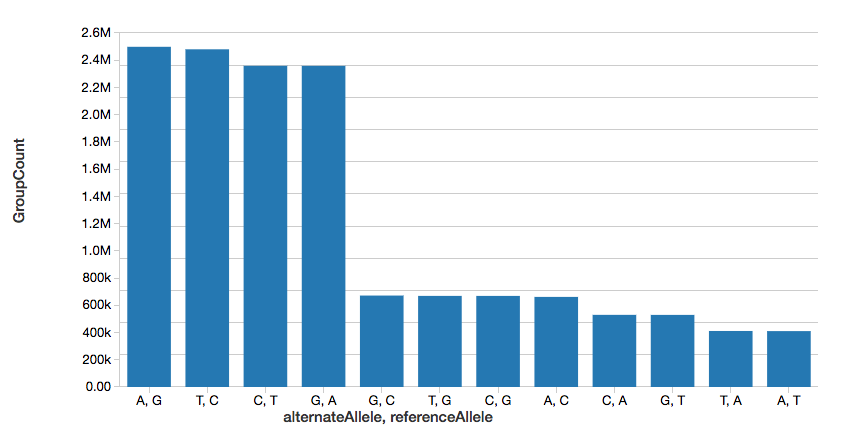
注意,显示命令是砖的工作空间的一部分,允许您查看DataFrame使用砖可视化(即不需要编码)。
变异数
因为我们带注释的变异和功能的影响,我们可以继续我们的分析通过观察我们看到变体的传播效果。大部分的变异检测到侧面区域编码蛋白质,这些被称为非编码变体。
显示器(exomes.groupBy (“mutationType”)。数())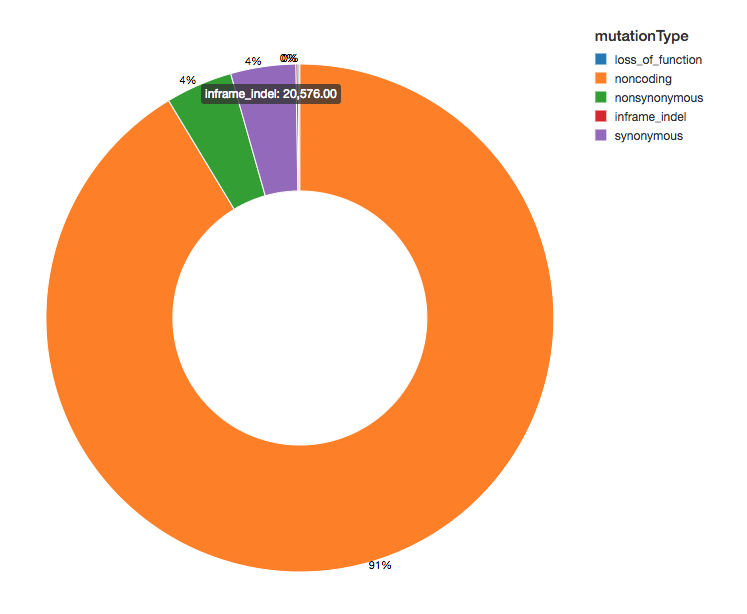
氨基酸替换的热图
继续我们的外显DataFrame,我们计算氨基酸替换下面的代码片段。类似于前面的DataFrames,我们将创建另一个动态DataFrame (aa_counts新的数据处理的),这样DataFrame外,随后将反映在氨基酸替换项。我们也写数据到内存中(即。.format(“记忆”)),每60年代(即加工批次。触发(processingTime = 60秒))所以下游熊猫的热图代码可以处理和可视化的热图。
#计算氨基酸替换编码= get_coding_mutations(外)aa_substitutions = get_amino_acid_substitutions (coding.select (“proteinHgvs”),“proteinHgvs”)aa_counts = count_amino_acid_substitution_combinations (aa_substitutions)aa_counts。\writeStream。\格式(“记忆”)。\queryName (“amino_acid_substitutions”)。\outputMode (“完整的”)。\触发(processingTime =“60秒”)。\start ()下面的代码片段读前面的amino_acid_substitutions火花表,确定最大计数,创建一个新的大熊猫从火花表数据透视表,然后情节的热图。
#使用熊猫和matplotlib来构建的热图amino_acid_substitutions=spark.read.table (“amino_acid_substitutions”)max_count=amino_acid_substitutions.agg(外汇。马克斯(“替换”))。收集()(0][0]aa_counts_pd=amino_acid_substitutions.toPandas ()aa_counts_pd=pd.pivot_table (aa_counts_pd值=“替换”、索引=(“参考”),列=(“替代”),fill_value=0)无花果、斧头=plt.subplots ()与sns.axes_style(“白”):斧头=sns。热图(aa_counts_pd,=max_count*0.4,cbar=假,annot=真正的,annot_kws={“大小”:7},fmt=“d”)plt.tight_layout ()显示(图)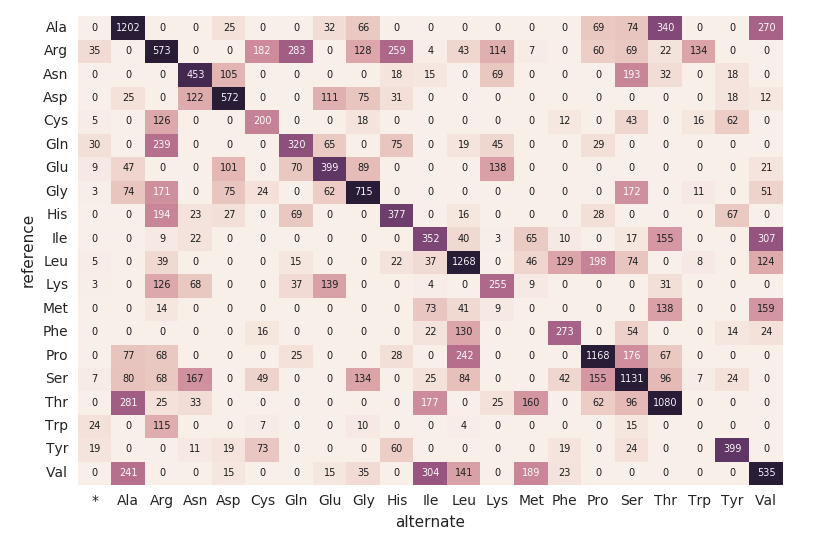
迁移到一个连续的管道
到目前为止,前面的代码片段和可视化表示一个竞选一个sampleId。但是因为我们使用结构化流和砖三角洲,这段代码可以使用(没有任何变化)构建一个生产数据管道计算质量控制统计数据不断,样品通过我们的管道。为了证明这一点,我们可以运行下面的代码片段,将加载整个数据集。
进口时间拼花=“dbfs: / databricks-datasets /基因组学/ annotations_etl_parquet /”文件= dbutils.fs.ls(铺)counter =0为样本在文件:计数器+ =1annotation_path = sample.pathsampleId = annotation_path.split (“/”)[4].split (“=”)[1]变量= spark.read。格式(“铺”).load (str(annotation_path))打印(“运行”+ sampleId)如果(sampleId ! =“SRS000030_SRR709972”):variants.write。格式(“δ”)。\模式(“添加”)。\保存(delta_stream_outpath)time . sleep (10)正如前面所描述的代码片段,的来源外显DataFrame文件加载到delta_stream_output文件夹中。最初,我们为一个加载一组文件sampleId(例如,sampleId = " SRS000030_SRR709972 ")。前面的代码片段现在需要所有生成的镶花的样品(即。拼花)和增量加载这些文件sampleId到相同的delta_stream_output文件夹中。下面的GIF动画展示的缩写输出前面的代码片段。
https://www.youtube.com/watch?v=JPngSC5Md-Q
想象你的基因组学管道
当你滚动回你的笔记本,你会注意到外显DataFrame现在自动加载新的sampleIds。因为我们的基因组学的结构化流组件管道连续运行,尽快处理数据加载到新文件delta_stream_outputpath文件夹中。通过使用砖三角洲的格式,我们可以确保事务一致性的数据流到DataFrame外。
https://www.youtube.com/watch?v=Q7KdPsc5mbY
而不是最初创建的外显DataFrame,注意结构化流媒体监控仪表板(即现在加载数据。,the fluctuating “input vs. processing rate”, fluctuating “batch duration”, and an increase of distinct keys in the “aggregations state”). As the外显DataFrame处理,注意的新行sampleIds(和变体数量)。这个动作也可以看到有关集团通过突变类型查询。
https://www.youtube.com/watch?v=sT179SCknGM
事务性一致与砖三角洲,任何新的数据在每一步我们的基因组学管道。这是很重要的,因为它确保您的管道是一致的(维护数据的一致性,即保证所有的数据是“正确的”),可靠(事务完全成功或失败),并可以处理实时更新(同时处理很多事务的能力和任何停机的故障不会影响数据)。因此即使我们下游的氨基酸替换的数据地图(有很多额外的ETL步骤)刷新无缝。
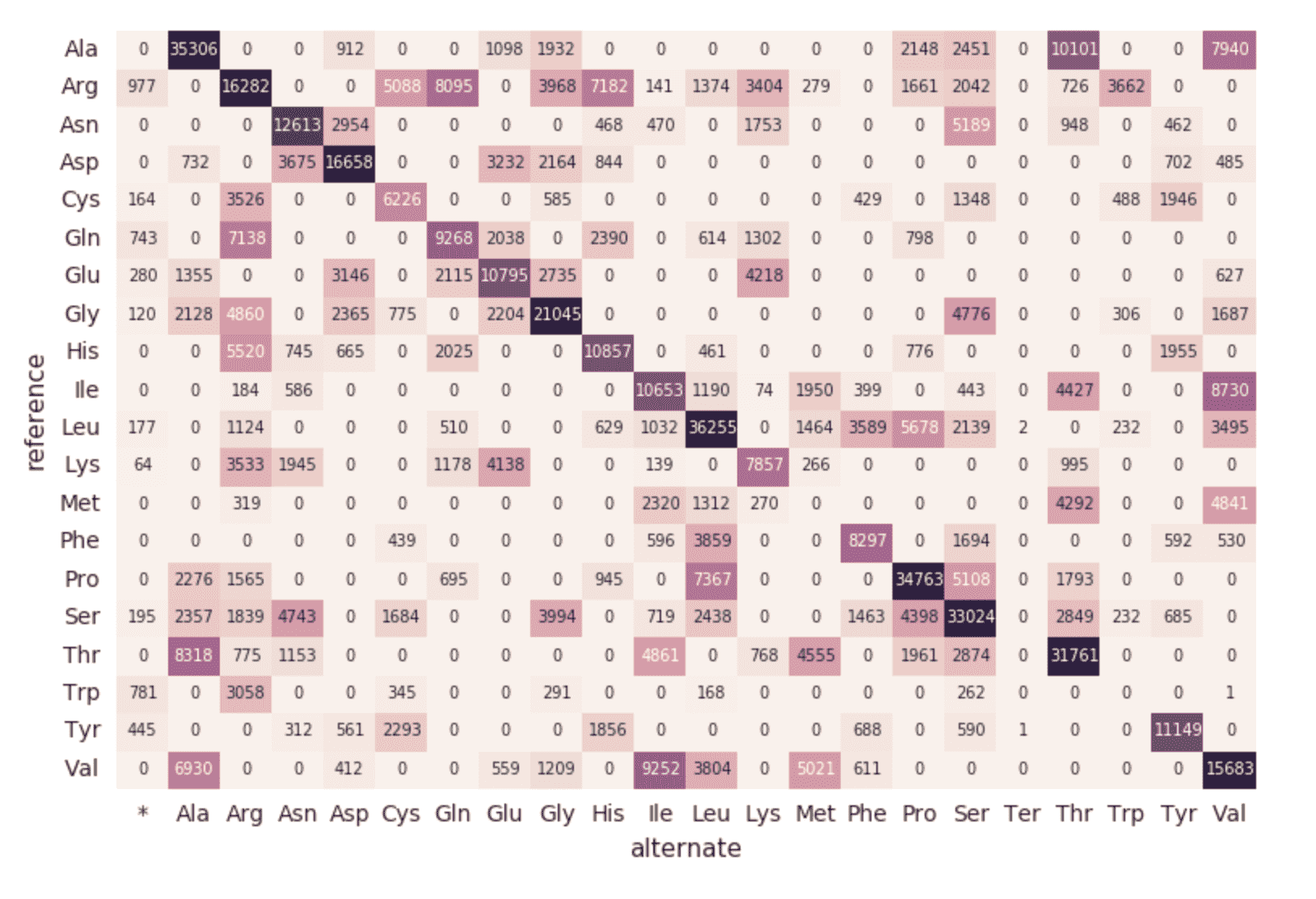
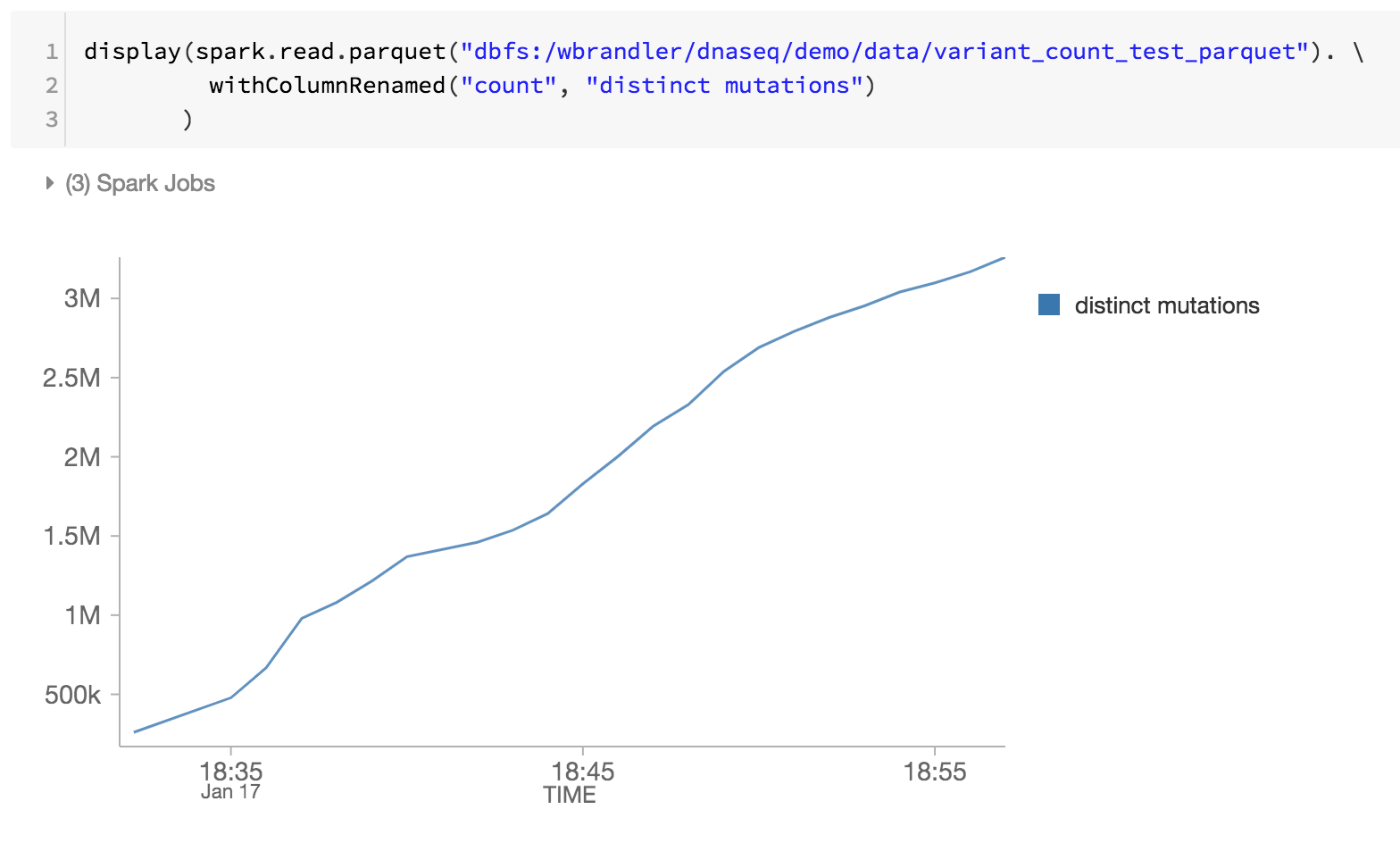
作为基因组学管道的最后一步,我们也监控不同的突变通过回顾砖三角洲DBFS内铺文件(即在一段时间内增加不同的突变)。
总结
使用砖统一分析平台的基础——与砖δ- bioinformaticians特别关bob体育亚洲版注和研究人员可以应用bob体育客户端下载分布式分析事务一致性砖统一为基因组学分析平bob体育亚洲版台bob体育客户端下载。这些抽象允许从业者简化基因组数据管道。在这里,我们已经创建了一个基因组样本质量控制管道不断随着新样本处理过程数据,无需人工干预。是否执行ETL或者执行复杂的分析,数据将流过你的基因组学管道迅速,没有中断。自己试试今天通过下载简化基因组大规模管道砖三角洲的笔记本。
开始分析大规模基因组学:
- 阅读我们的统一为基bob体育亚洲版因组学分析解决方案指南
- 下载简化基因组大规模管道砖三角洲的笔记本
- 注册一个免费试用砖统一为基因组学分析bob体育亚洲版
确认
由于永胜黄和迈克尔·奥尔特加的贡献。
访问在线三角洲湖中心要了解BOB低频彩更多,请下载最新的代码,并加入三角洲湖社区。