
介绍GlowGR:一种工业规模、超快速和敏感的遗传关联研究方法








2020年6月25日 在公司博客上
今天,我们宣布,作为Glow项目的一部分,我们正在为开源生物信息学社区提供一种新的全基因组回归方法。bob下载地址
大量具有配对临床和基因组序列数据的个体能够前所未有地深入了解人类疾病生物学。人口研究,比如英国生物库,基因组学英格兰,或亚洲基因组10万数据集正在推动对基因数据处理方法创新的需求。这些方法包括全基因组关联研究(GWAS),它丰富了我们对疾病遗传结构的理解,并用于尖端工业应用,如鉴定药物开发的治疗靶点.然而,这些数据集提出了新的统计和工程挑战。诸如SAIGE和Bolt-LMM等工具已经解决了统计上的挑战,但它们很难建立,并且在生物银行规模的数据集上运行速度非常慢。
在典型的GWAS中,单个表型(生物体的可观察特征),如胆固醇水平或糖尿病诊断状况,被测试与基因组中数百万个遗传变异的统计关联。复杂的混合模型和基于全基因组回归的方法已经开发出来,用于在测试遗传关联时控制大型遗传研究人群固有的相关性和群体结构;有几种方法,例如BOLT-LMM,SAIGE,fastGWA在生物银行规模的项目中,使用一种称为全基因组回归的技术来敏感地分析单个表型。然而,深度表型生物库规模的项目可能需要数万个单独的GWASs来分析临床变量的全谱,而且目前的工具在大规模运行时仍然非常昂贵。为了应对有效分析此类数据集的挑战,Regeneron遗传学中心刚刚开发了一种全基因组回归方法的新方法,可以同时在数百种以上的表型中运行GWAS。这个令人兴奋的新工具提供了与当前最先进的方法相同的卓越测试能力,而计算成本仅为其一小部分。
这种新的全基因组回归(WGR)方法将全基因组回归问题重铸为许多小的、遗传区域特定模型的集成模型。此方法在章节中描述预印本今天发布,并在c++工具regenie.作为Regeneron遗传学中心和Databricks在开源方面合作的一部分bob下载地址项目发出在此,我们激动地宣布GlowGR,这是一种闪电般快速且高度可扩展的WGR算法分布式实现,由Apache Spark™从头开始设计,并与其他Glow功能集成。使用GlowGR,可以在几分钟内同时完成对数十种表型的WGR分析,而使用现有的最先进工具需要数百或数千小时才能完成这项任务。此外,GlowGR沿样本和遗传变异矩阵维度分布,允许线性缩放和高度的数据和任务并行。GlowGR无缝插入任何现有的GWAS工作流程,以可忽略不计的计算成本立即提高关联检测能力。
利用全基因组回归实现高精度和高效率
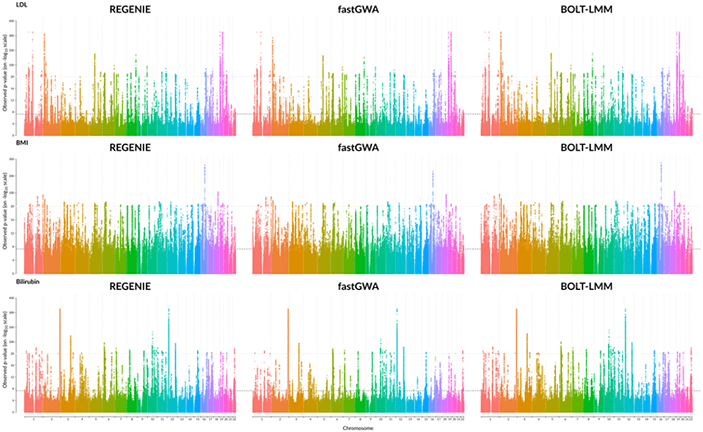
这种全基因组回归工具有很多优点。首先,它更有效:正如在单节点,开源regenie工具,全基因组回归比SAIGE、Bolt-LMM或fastGWA快几个数量级,同时产生等效的结果(图1)。其次,并行化很简单:在下一节中,我们将描述如何在开源项目Glow中使用Apache Spark实现全基因组回归。
除了性能考虑外,全基因组回归方法产生的协变量与标准的GWAS方法兼容,并且消除了传统方法中由群体结构引起的虚假关联。下面图2中的曼哈顿图比较了使用标准协变量的传统线性回归GWAS与使用WGR生成的协变量的线性回归GWAS的结果。与现有的GWAS工具相比,GlowGR的这种灵活性是另一个巨大的优势,它将允许对已经在Glow中可用的关联测试框架进行各种令人兴奋的扩展。
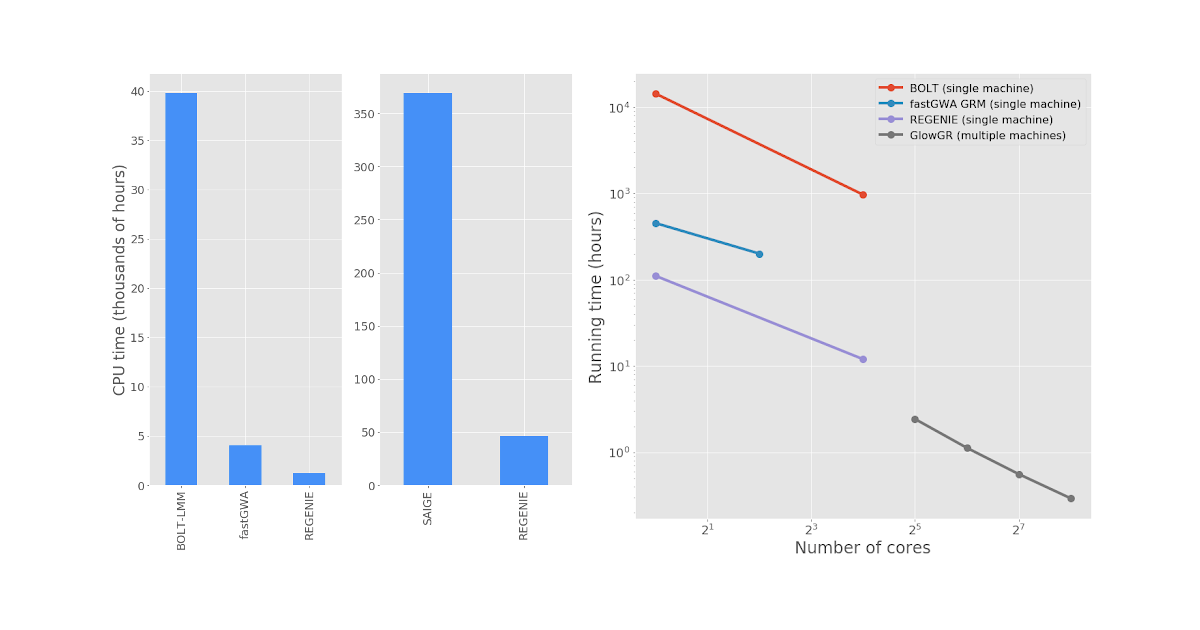
图3显示了GlowGR、REGENIE、BoltLMM和fastGWA之间的性能比较。我们对Glow中实现的全基因组回归测试与c++中可用的实现进行了基准测试单节点regenie工具验证了该方法的准确性。我们发现这两种方法在统计上取得了相同的结果。我们还发现,Glow中基于Apache Spark™的实现与所使用的节点数量成线性关系。
在Project Glow中缩放全基因组回归
使用GlowGR执行WGR分析有5个步骤:
- 将基因型矩阵分成相邻的SNPs块(每个块约1000个SNPs,称为位点)
- 在每个基因座内拟合多个脊模型(~10个)
- 使用产生的脊模型将轨迹从1000个特征的矩阵减少到10个特征(每个特征是一个脊模型的预测)
- 将所有位点的结果特征汇集到一个新的简化特征矩阵X中(每个位点由L个位点X个脊模型组成N个个体)
- 拟合X的最终模型,以获得全基因组对表型Y的贡献。
Glow提供了如图4所示的易于使用的抽象,用于将大型基因型矩阵转换为阻塞矩阵(下图,左),然后拟合全基因组回归模型(下图,右)。中加载的数据可以应用这些Glow理解的基因型文件格式,包括VCF, Plink和BGEN格式,以及存储在Apache Spark™原生文件格式中的基因型数据三角洲湖.

Glow为数量性状提供了WGR方法的实现,一个二元性状变体正在进行中。由GlowGR创建的协变量调整表型可以写成一个Apache拼花™或三角洲湖数据集,可以很容易地加载和分析Apache火花,熊猫,以及其他工具。最终,在全基因组关联研究中使用WGR计算的协变量就像运行下面图5所示的命令一样简单。该命令由Apache Spark™在所有被测基因标记之间并行运行。

加入我们,在Glow中尝试全基因组回归!
全基因组回归可在发光,这是一个开源项目bob下载地址托管在Github上他拥有Apache 2许可证。你可以从这个笔记本说明如何使用GloWGR,请阅读预印,通过阅读我们的项目文档,或者你可以创建存储库的一个分支今天就开始贡献代码。Glow已安装在Databricks Genomics运行时(Azure|AWS你今天就可以开始预演了。